VariantDB
UZA-USERS :All data of UZA accounts has been migrated & accounts were disabled here!Use : https://variantdb.uza.be/'
Documentation : Example Filtering Strategies
VariantDB allows filtering according to several strategies. Below are a few example setups.
1. De Novo Trio:
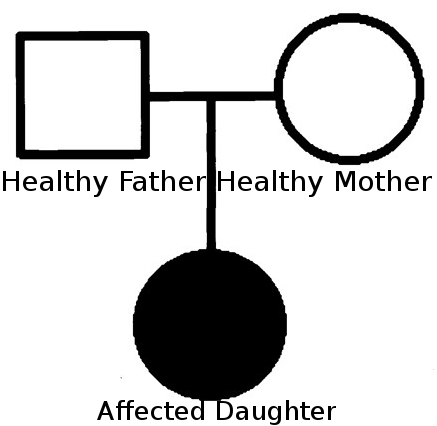
- Case: Two healthy parents, no familial history. Expected model: De novo mutation. Sequencing data from all three indivuals is available.
- Strategy: Select variants not present in parents, with effect on protein function
- Upload the three VCF (and BAM files to variantDB (see here for instructions)
- Set Family relations in the Sample Management section: Assign obht parents to the offspring sample (see image below)
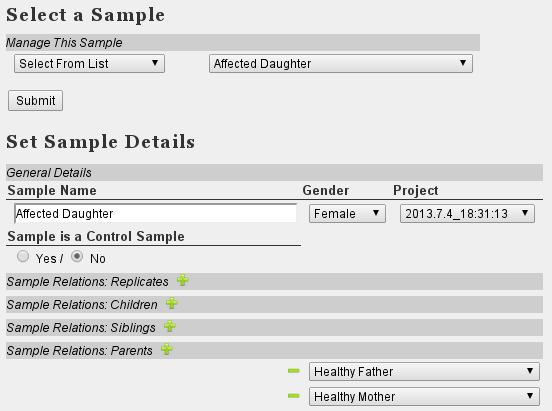
- At 'Variant Filter', select the correct sample 'Affected Daughter'
- Set the following filters:
- Filter On Family Information : 'Not Match', 'In Parent', 'Father + Mother', 'As Any Genotype'
- Filter On snpEff Information : 'Match', 'Effect Impact', 'High'
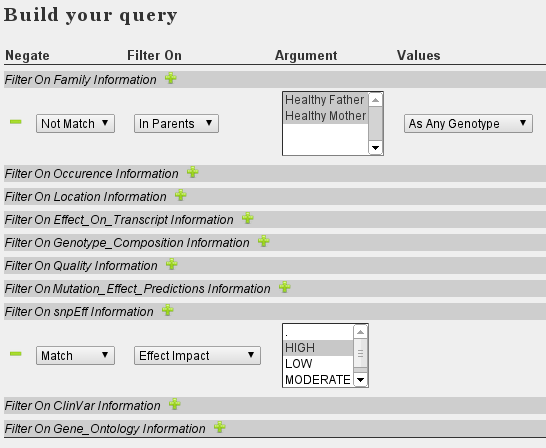
- Select some relevant annotations
- Execute Query
The above approach is a good starting point selecting high impact variants that are not present in the parents. We excluded all variants present in the parents, regardless of the exact genotype (heterozygous/homozygous) in either child or parents. The resulting variants have a severe impact (frameshift/stopgain/...) on at least one transcript as annotated by snpEff. To further restrict your result, you might add some quality thresholds. If no clear candidate remains, adjust your filters. For example: add 'moderate' effect impact from snpEff. Alternatively, remove the snpEff filter and activate filtering on CADD score, for example with a PhredScaled CADD score above 30. This relates to the 0.1% most likely pathogenic SNVs of all SNVs possible in the genome.
2. Autosomal Dominant Family:
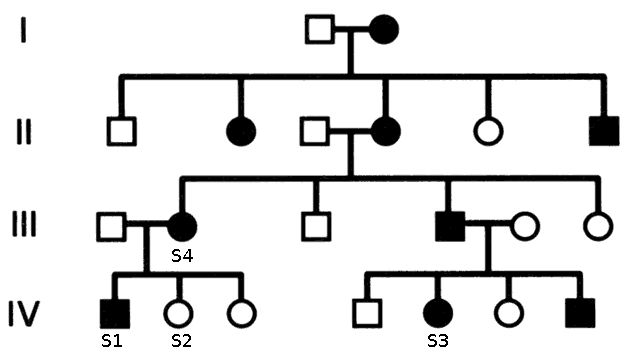
- Case:Large pedigree with familial history. Expected model: Automsomal Dominant. Sequence data from three affected family members and one unaffected sibling (S2) of the index patient (S1).
- Strategy Select variants shared amongst affected members, absent from healthy sibling
- Set Family relations. Although Cousin is not a direct option in VariantDB, you can assign S3 as a sibling of S1 without any other consequences
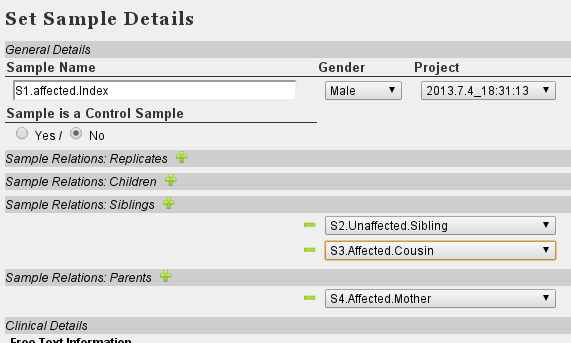
- At 'Variant Filter', select the correct sample 'S1.Affected.Index'
- Set the following filters:
- Filter On Family Information : 'Match', 'In Parent', 'S4.Affected.Mother'
- Filter On Family Information : 'Match', 'In Sibling', 'S3.Affected.Cousin'
- Filter On Family Information : 'Not Match', 'In Sibling', 'S2.Unaffected.sibling'
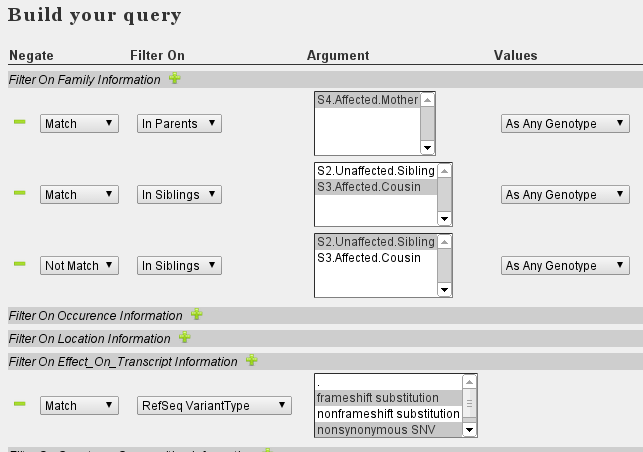
- Select Filters with regard to function (eg RefSeq, snpEff, CADD, ...)
- Select Annotations
- Execute Query
The above approach selects variants shared amongst affected family members and not present in healthy family members. As we expect dominant inheritance, no criteria are set on the genotype, as any genotype (heterozygous or homozygous) causes the phenotype.
3. Autosomal Recessive
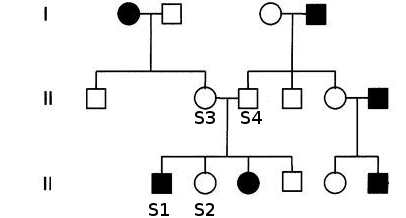
- Case:Pedigree with familial history. Expected model: Automsomal Recessive. Sequence data from affected index patient (S1), both parents (S3-4) (and unaffected sibling (S2)).
- Strategy Select variants homozygous in index, heterozygous in both parents (and not homozygous in sibling)
- Set Family relations.
- At 'Variant Filter', select the correct sample 'S1.Affected.Index'
- Set the following filters:
- Filter On Family Information : 'Match', 'In Parent', 'S4.Mother', 'Heterozygous'
- Filter On Family Information : 'Match', 'In Sibling', 'S3.Father', 'Heterozygous'
- Filter On Family Information : 'Not Match', 'In Sibling', 'S2.Unaffected.sibling', 'Homozgyous'
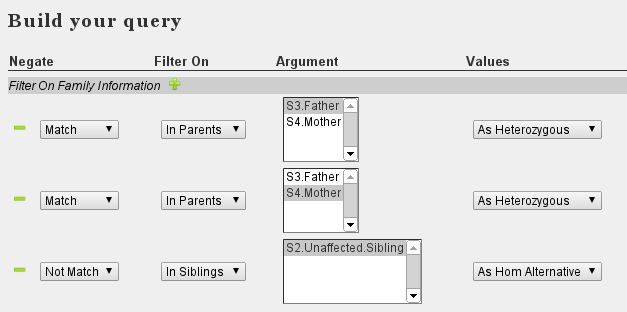
- Select Filters with regard to function (eg RefSeq, snpEff, CADD, ...)
- Select Annotations
- Execute Query
Note the difference with the De novo filtering. For de novo variants, we set that variants in the index patient should not be present in either father or mother. This was combined in a single statement (multi-select box, not match : Translates to Not in Father OR Mother). In contrast, to ensure that the variant is present in both father and mother we need to set two rules, each specifying to match one parent. Furthermore, we specified the genotype to match.
IMPORTANT: Selecting multiple values in a single filtering rule matches if one of the values matches (eg: either non-synomous, or frameshift or stopgain).
4. Linkage Based
- Case:Pedigree with familial history. SNP-array based linkage analysis resulted in a single region with high LOD score. Sequencing data from one affected indivual
- Strategy Select variants within the linkage region
- At 'Variant Filter', select the correct sample
- Set the following filters:
- Filter On Location : 'Match', 'Chromosome', 'Chromosome of linkage region'
- Filter On Location : 'Match', 'Position', 'Bigger than', 'linkage region start coordinate'
- Filter On Location : 'Match', 'Position', 'Smaller than', 'linkage region end coordinate'
- Select Filters with regard to function (eg RefSeq, snpEff, CADD, ...)
- Select Annotations
- Execute Query
This filtering method allows to select variants specific to a genomic region. If additional samples from the linkage study are available, results can be refined using the above described family based filters.
5. Complex pedigree or Case/Control study
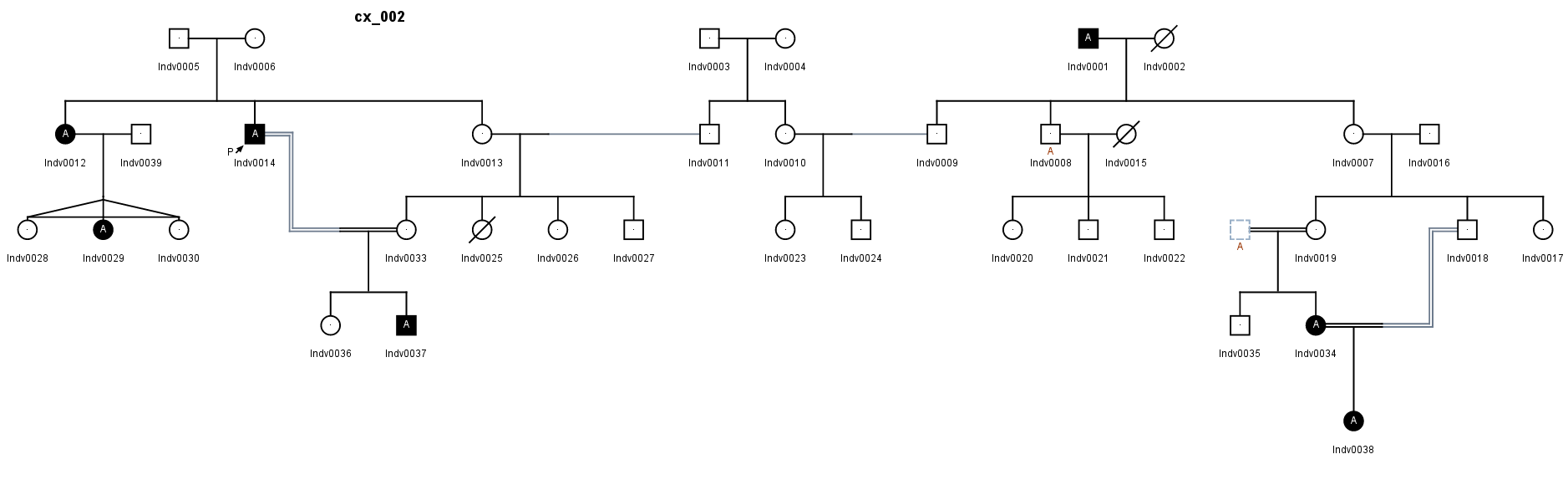
- Case:Large Cohort or family screened. Expected inheritance : complex or incomplete penetrance.
- Strategy Select variants with minimal occurence ratio in cases, and maximal occurence ratio in controls
- Order your affected in unaffected samples into two seperate projects from the sample management page. In this case:
- Project Complex_Pedigree_Affected : Individuals 12, 14, 37, 1, 34 and 38
- Project Complex_Pedigree_Unaffected : All other available individuals
- At 'Variant Filter', select an affected sample (eg. Indv0001)
- Set the following filters:
- Filter On Occurence : 'Match', 'Relative Occurence By Project', 'Cases_Project', 'Bigger Than', '0.83'. This allows one false-negative amongst the 6 affected samples.
- Filter On Occurence : 'Match', 'Relative Occurence By Prroject', 'Control_Project', 'Smaller Than' '0.1'. This allows healthy family members to carry the mutation. Adjust this value to reflect the degree of penetrance.
- Select Filters with regard to function (eg RefSeq, snpEff, CADD, ...)
- At 'Filter Logic', further refine the filtering scheme if necessary. As shown in the example below, you might want to bypass quality filters, if a mutation is known in ClinVar to be pahogenic. As an example, the scheme now returns all high quality variants highly represented in affected family members and rare among the healthy siblings. In addition, it will return known pathogenic mutations from ClinVar, and high quality variants with a CADD-score over 40, which are also very likely to be pathogenic, regardless of frequencies in the pedigree.

- Select Annotations
- Execute Query
Here we say that 90% of the affected family members should harbour the mutant allele, while at most 10% of unaffected family members may harbour it. Setting the affected-ratio to less than 100% allows for false negative calls, while setting a maximal ratio for unaffected family members allows for incomplete penetrance. This filtering approach is also available using absolute occurence numbers, when the pedigree is too small for frequency measures. In case of large case/control cohorts, a similar approach with adequate frequencies can be applied.