VariantDB
UZA-USERS :All data of UZA accounts has been migrated & accounts were disabled here!Use : https://variantdb.uza.be/'
Documentation : IGV
1. Installing IGV
Download the IGV from the following website: Download. This will install the BETA version of IGV, which might give some problems. However, since VariantDB switched to CRAM as its storage backend, using the BETA version is mandatory. Once the main IGV release supports CRAM files, we'll update this link.
2. Adding our Reference Genome
The Biomina/medgen galaxy server uses a cleaned up version of the UCSC genome build to convert BAM files to CRAM format. In particular, all chrXX_un and chrXX_random entries were removed. If the specified reference in the VCF/BAM file is not recognized as the same version, imported files are kept as BAM files.
It is highly recommended to download and install this reference genome locally, as it will significantly improve IGV performance. This is done using the following steps, and is only needed once, as IGV will remember these settings:
- Download the reference genome HERE
- Unzip the file. It will require approximately 3.5Gb of space. You can delete the zip file afterwards.
- Launch IGV
- From the file menu, select 'Load Genome from file'
- Browse to the folder you extracted the zipfile in, and select 'hg19.variantdb.genome'
- Press OK and wait a minutes
- If it works, you should now have the 'Human hg19 (VariantDB-CRAM)' entry in the dropdown list on the top left of the screen

3. Get Sample Data into VariantDB
To load data into IGV from the VariantDB itself, there are multiple options. First, it can be imported from our Galaxy server using a dedicated tool on the Biomina Galaxy instance, following these steps:
- Use galaxy on this location
- Analyze your sample, and call SNP/indels by GATK Unified genotyper. This is currently the only supported format.
- Select the "Send VCF file to the database" tool (under "Our Tools").
- Provide the GATK VCF file from the first drop-down list
- Select "Yes" in the second field to Store VCF and BAM files. This will send both files to the VariantDB storage location
- Finally, provide a BAM file and a sample name. The sample name and gender can be adapted later in VariantDB
Second, it can be directly imported using the Web-UI Instructions are listed on there. Third, data can be imported using API. This is documented here.

4. Load Sample Data into IGV
If the datafiles were sent to VariantDB from galaxy, you will now see a link on the top right of the "Variant Filter" page, next to the sample you have selected. Depending on what is available (VCF, BAM or both), this link will differ. Click on the link to load the data into a running IGV session, so make sure IGV was already started. As only fragments of data are loaded when visualized, this should go rather fast./p>
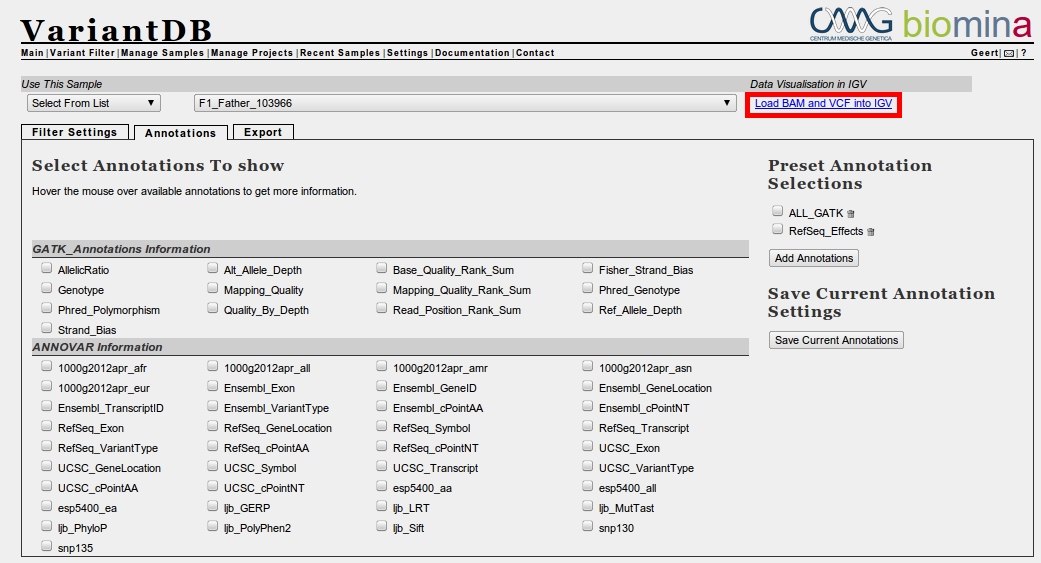
5. Navigating IGV through VariantDB
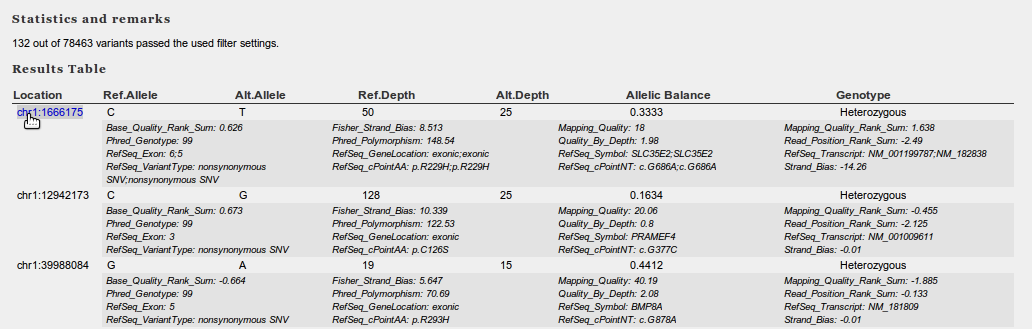
Results returned from the filter are presented in a tabular fashion. The first field in this table is always the genomic location of the variant. This location is a hyperlink to IGV. By clicking on the link, the display of IGV will jump to the location (+ 20bps on each side), and if present, the corresponding reads will be loaded from the BAM file.